Diabetes Liver Disease – The Burden
Nonalcoholic fatty liver disease (NAFLD) and type 2 diabetes (T2DM) often coexist. NAFLD includes a spectrum of pathological conditions, which range from simple steatosis (NAFL), nonalcoholic steatohepatitis (NASH), cirrhosis and hepatocellular carcinoma.1
The high prevalence rates of NAFLD have paralleled the rapidly progressing epidemic of obesity and T2DN over the past two decades. In fact, at the intersection of related risk factors, epidemiology, and pathophysiology, NAFLD and T2DM exist. In terms of precedence, NAFLD is relatively recent in its recognition as a major chronic condition compared to T2DM. Compared to the occurrence of NAFLD without chronic hyperglycemia, the co-existence of NAFLD and T2DM greatly increases the risk of developing NASH and cirrhosis. NAFLD’s participation as an independent indicator of events involving cardiovascular disease (CVD) remains debatable.2
Interestingly, the highest mortality in NAFLD is related not to end-stage liver disease or HCC risk, but to a deteriorating risk profile of CVD that may be due to T2DM comorbidity and other proven risk factors of CVD.1
Spectrum and Pathophysiology
Overweight and obesity are the main drivers of metabolic diseases and NAFLD. Fat distribution, adipose tissue (AT) functionality and IR constitute the basis of metabolic disturbances such as MetS, diabetes and NAFLD.3
From Diabetes to NAFLD/NASH
T2D results from an imbalance between insulin sensitivity and insulin secretion. In the progression from normal to impaired glucose tolerance to T2D, insulin secretion increases to overcome insulin resistance. At the same time there is a decrease in insulin clearance (mainly hepatic), especially in individuals with NAFLD, that determines higher peripheral insulin concentrations. However, despite high concentrations, insulin secreted is insufficient (beta-cell dysfunction) and therefore there is an increase in both fasting and postprandial glucose concentrations. Subjects become T2D only when the beta cells are unable to increase insulin secretion (beta-cell failure) and overcome peripheral IR the physiological mechanisms underlying the development of NAFLD are mainly the alterations in glucose and lipid metabolism, insulin resistance (IR) and insulin secretion, explaining the close association between NAFLD and T2D.4
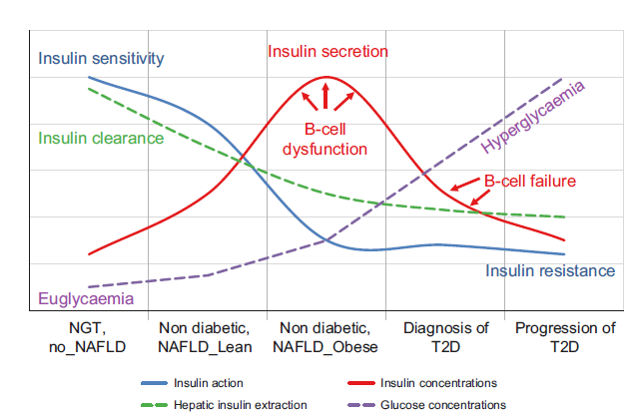
Figure 1: Natural history of T2D in the context of NAFLD4
From NAFLD/NASH to Diabetes
Increased adiposity, as often found in NAFLD and T2D, is associated with adipocyte IR and dysfunction. This results in excess FFA release into the blood stream and predisposes to lipotoxicity, i.e. when FFA overflow from adipose tissue leads to excess lipid uptake by tissues such as the liver, pancreas or muscle. Lipotoxicity is associated with increased peripheral IR, hepatic glucose production and gluconeogenesis leading to hyperglycaemia, as well as pancreatic beta-cell dysfunction with impaired insulin secretion. Both lipotoxicity and glucotoxicity contribute to insulin resistance, ectopic fat accumulation and beta-cell dysfunction and vice versa. In NAFLD, lipotoxicity and insulin resistance have been recognised as pathophysiological mechanisms responsible of development and progression to a more severe form of this disease. T2D is a chronic condition of glucotoxicity, although also lipotoxicity is often present and are responsible not only of insulin resistance but also of impaired insulin secretion.4
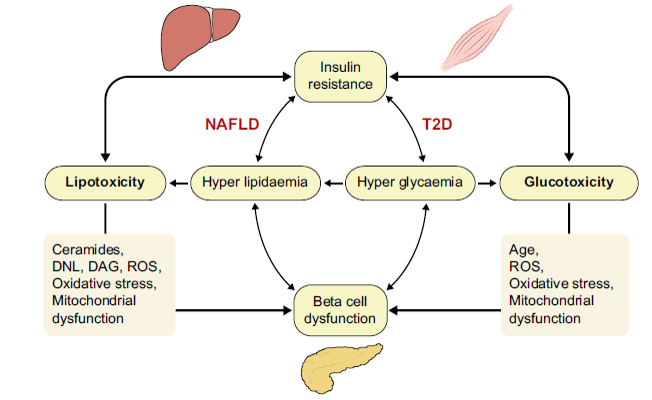
Figure 2: Relationship between lipo- and glucotoxicity,
insulin resistance and beta-cell function. DAG, diacylglycerol; DNL, de novo lipogenesis; NAFLD, non-alcoholic fatty liver disease; ROS, reactive oxygen species4
Guideline Recommendations
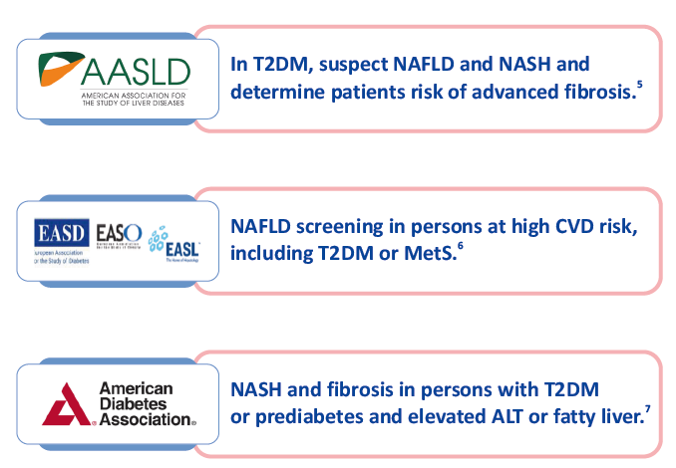
Current Management Challenges
In NASH
There is a lack of effective therapies that are specifically approved for the treatment of Advanced Fibrosis due to NASH, including the reversal of fibrosis. Thus, a high unmet need remains for effective treatments that halt progression of fibrosis and prevent cirrhosis. New treatment options are therefore needed to halt the progression of Advanced Fibrosis due to NASH to prevent the onset of cirrhosis and its associated costs, HCC, or end-stage liver disease.8
In PBC
A major landmark in the history of PBC was the introduction of ursodeoxycholic acid (UDCA) as an effective therapy. After many years of controversy, there is now a consensus to accept UDCA as the standard therapy. UDCA has been shown to extend transplantation–free survival, especially when started early in the course of the disease. About 30% to 40% of patients will not have an optimal biochemical response to UDCA. These patients exhibit more rapid disease progression than those with normalisation of serum alkaline phosphatase (ALP), aminotransferases and bilirubin. In a meta-analysis of 4,845 patients with PBC, the strongest predictors of death or liver transplantation were ALP levels more than 1.5–3x the upper limit of normal range and abnormal serum bilirubin.9
By contrast, an estimated 1 in 3 PBC individuals reveals UDCA suboptimal or non-response as assessed by variable biochemical surrogate endpoints with a special emphasis on serum alkaline phosphatase (ALP) activity, thus conferring high risk of continued disease progression despite treatment (“high risk PBC”).10
Historically, budesonide was the first second-line treatment in PBC to show promising results in association with UDCA. Importantly, budesonide is inadvisable in cirrhotic patients in whom its first-pass effect is impaired, exposing them to significantly higher plasma concentrations and increased risk of systemic side effects.9
Farnesoid X-Receptor and its Role in Health & Diseases
Farnesoid X receptor (FXR), a metabolic nuclear receptor, plays critical roles in the maintenance of systemic energy homeostasis and the integrity of many organs, including liver and intestine. It regulates bile acid, lipid, and glucose metabolism, and contributes to inter-organ communication, in particular the enterohepatic signaling pathway, through bile acids and fibroblast growth factor-15/19 (FGF-15/19). The metabolic effects of FXR are also involved in gut microbiota. In addition, FXR has various functions in the kidney, adipose tissue, pancreas, cardiovascular system, and tumorigenesis. Consequently, the deregulation of FXR may lead to abnormalities of specific organs and metabolic dysfunction, allowing the protein as an attractive therapeutic target for the management of liver and/or metabolic diseases.11
FXR: An evolving therapeutic target in diabetic liver disease
In the liver, FXR is expressed primarily in hepatocytes, Kupffer cells, and endothelial cells. FXR activation has pleiotropic effects on NASH including increased glucose disposal, even in type 2 diabetic patients, and decreased lipogenesis along with decreased inflammation. Together, these reduce fibrosis and complement direct antifibrotic effects on hepatic stellate cells. Through these effects, FXR hits key elements of the NASH cascade and has a strong biological rationale for use.12
Obeticholic acid (OBIHOLIC): A Paradigm shift in the management of PBC & NAFLD/NASH
Historically, ursodeoxycholic acid (UDCA), a naturally occurring bile salt, was the only approved therapy for PBC. However, 40% of PBC patients do not respond to UDCA. Therefore, a large percentage of patients have disease progression to end-stage liver disease. Many other agents have been trialed in treating this puzzling disease, including colchicine, fibrates, and methotrexate. However, these other therapies have not proven to be effective. Thus, there was a critical need for new pharmacotherapies to treat PBC and prolong survival from this disease. 13 Obeticholic acid (OCA) is a new treatment for PBC that has received approval by the Food and Drug Administration (FDA).1 OCA is a semi-synthetic hydrophobic BA analogue, which is a highly selective agonist of FXR, with activation potency similar to the endogenous BA chenodeoxycholic acid but is 100 times more potent. The resulting beneficial effects of OCA on glucose and lipid metabolism and particularly hepatic inflammation make it a potential candidate for pharmacological treatment of a variety of conditions including PBC and NASH.14
Clinical development
OCA is the only therapy licensed by the FDA, EMA and endorsed by NICE as the second line therapy for PBC. Subsequent clinical trials have confirmed the efficacy of OCA in PBC, and have led to it becoming a licensed treatment as the second line therapy in USA and Europe. Further long-term studies are in progress in PBC to confirm a survival benefit. In addition,OCA treatment has shown encouraging results in NASH with a reduction in liver fibrosis.14
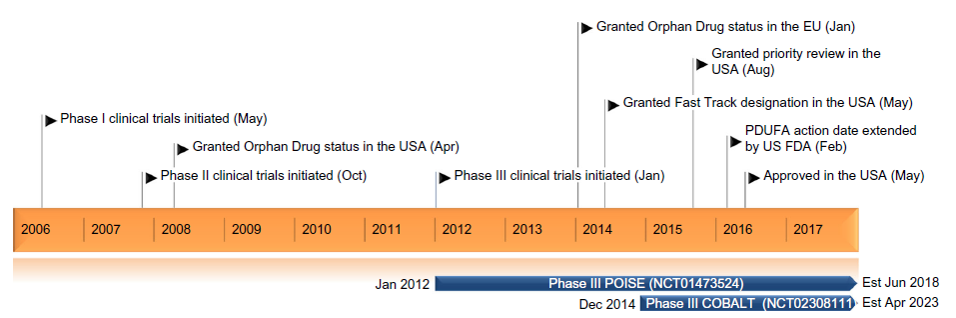
Figure 3: Key milestones in the development of obeticholic acid for the treatment of PBC15
Pharmacology
About OCA
Chemically, OCA is 3α,7α -dihydroxy- 6α -ethyl-5β-cholan-24-oic acid. It is a white to off-white powder. It is soluble in methanol, acetone and ethyl acetate. Its solubility in water is pH dependent. It is slightly soluble at low pH and very soluble at high pH. Its chemic al formula is C26H44O4, the molecular weight is 420.63 g/mol and the chemical structure is as shown in figure 4.16
Figure 4: Chemical structure of OCA16
Mechanism of action of OCA
OCA is a first-in-class agonist that selectively binds to the FXR. OCA works directly and indirectly to suppress bile acid production in the liver and increase bile flow, leading to reduced exposure of the liver to toxic levels of bile acids.13 Activation of FXR inhibits de novo bile acid synthesis from cholesterol in hepatocytes and increased clearance of bile acids from the hepatocytes. This results in the reduction of the overall bile acid pool, which protects against the toxic effects of the accumulation of bile acids, thus reducing hepatic exposure to bile acids.17
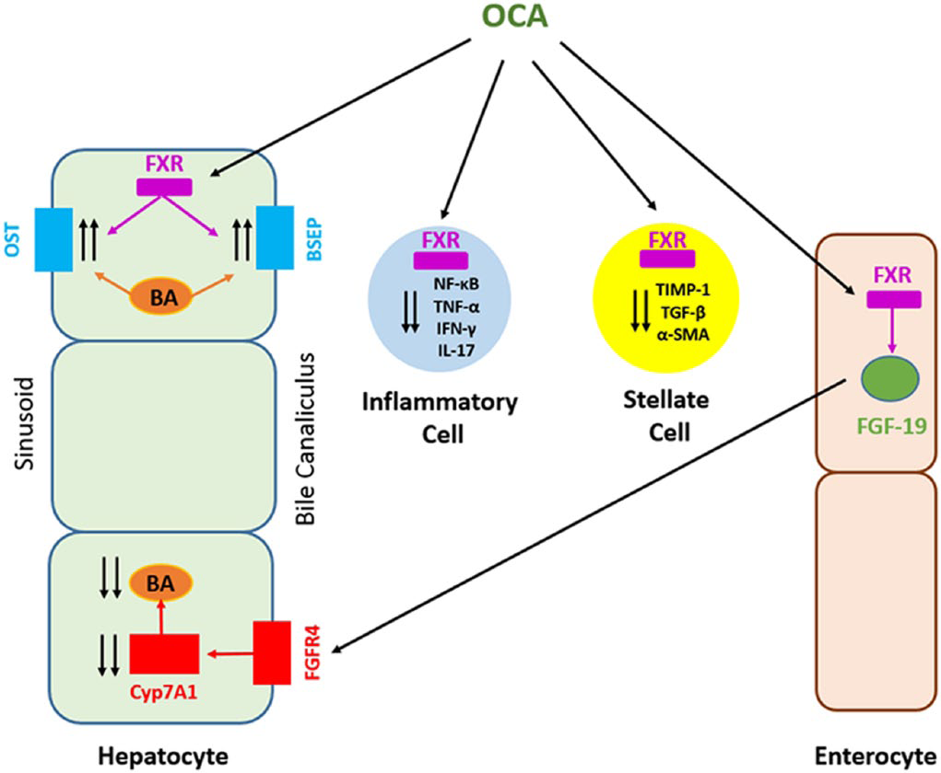
Figure 5: Mechanism of action of OCA17
Pharmacodynamic properties
Upon oral administration, OCA functions as a bile acid and signaling molecule in several pathways modulated by FXR. Also, OCA treatment modulated the expression of genes regulating lipid and glucose metabolism. Direct anti-inflammatory, anti-fibrotic and immune-modulatory effects of OCA were demonstrated in test systems such as cultured human hepatic stellate cells, mouse primary hepatocytes and human immune cells. Several safety pharmacology studies have evaluated the neurological, cardiovascular, pulmonary and gastrointestinal effects of OCA. Oral administration of OCA, up to 120 mg/kg, did not produce any apparent neuro-pharmacological or toxicological effects, changes in pulmonary function or GI propulsion, or biologically relevant effects on body temperature.18
Pharmacokinetic properties
Absorption: Following multiple oral doses of OCA 10 mg once daily, peak plasma concentrations (Cmax) of OCA occurred at a median time (Tmax) of approximately 1.5 hours.16
Distribution: Human plasma protein binding of OCA and its conjugates is greater than 99%. The volume of distribution of OCA is 618 L.16
Metabolism: After daily administration of OCA, there was accumulation of the glycine and taurine conjugates of OCA, which have in vitro pharmacological activities similar to the parent drug, OCA. The metabolite-to-parent ratios of the glycine and taurine conjugates of OCA were 13.8 and 12.3 respectively, after daily administration.16
Excretion: After administration of radiolabeled OCA, about 87% of the dose was excreted in feces through biliary secretion. Less than 3% of the dose was excreted in the urine with no detection of OCA.16
OBIHOLIC: Key Clinical Evidences
OCA as Monotherapy: FLINT Trial
Neuschwander-Tetri BA, et al., conducted a study to assess the efficacy of obeticholic acid in adult patients with NASH. The Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment (FLINT) trial was a multicentre, randomised trial of 72 weeks. 283 patients were randomly assigned 1:1 using a computer-generated, centrally administered procedure, stratified by clinical centre and diabetes status. The primary outcome measure was improvement in centrally scored liver histology defined as a decrease in NAFLD activity score by at least 2 points without worsening of fibrosis from baseline to the end of treatment.19
Results observed were: 19
- 45% of patients in the OCA group had improved liver histology (2-point or greater improvement in NAFLD activity score without worsening of fibrosis) compared with 21% in the placebo group (relative risk 1·9, 95% CI 1·3–2·8; p=0·0002)
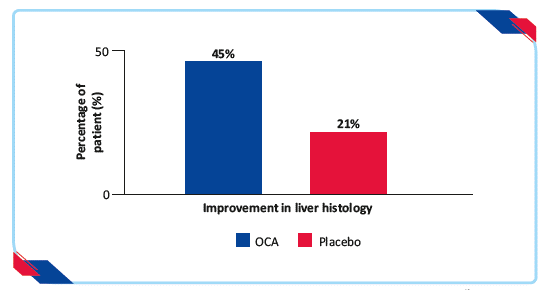
Figure 6: Proportion of patients achieving improvement in liver histology19
- Greater number of patients assigned to OCA had improvement in fibrosis, hepato cellular ballooning, steatosis, and lobular inflammation
- Greater mean change in the NAFLD activity score in patients treated with OCA than placebo (change from baseline=–1·7 vs –0·7; p<0·0001)
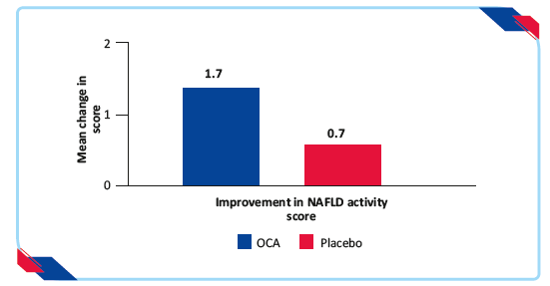
Figure 7: Proportion of patients achieving improvement in NAFLD activity score19
- Significant reductions in serum alanine aminotransferase and aspartate aminotransferase concentrations developed over the first 36 weeks of treatment with OCA and were sustained for the duration of treatment
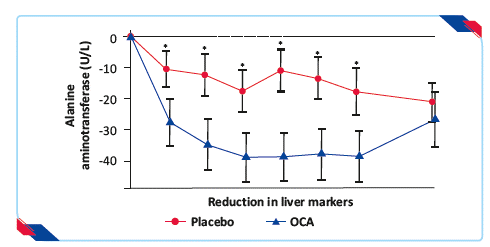
Figure 8: Proportion of patients achieving reduction in liver markers* p<0.0519
OCA improved the histological features of NASH, but its long-term benefits and safety need further clarification.19
OCA as Monotherapy: REGENERATE Trial
Younossi et al., in a multicentre, randomised, double-blind, placebo-controlled study, adult patients with definite NASH, NAFLD activity score of at least 4, and fibrosis stages F2–F3, or F1 with at least one accompanying comorbidity, were randomly assigned using an interactive web response system in a 1:1:1 ratio to receive oral placebo, OCA 10 mg, or OCA 25 mg daily. The primary endpoints for the month-18 interim analysis were fibrosis improvement (≥1 stage) with no worsening of NASH, or NASH resolution with no worsening of fibrosis, with the study considered successful if either primary endpoint was met.20
Results observed were:20
- Fibrosis improvement endpoint was achieved by 12% patients in the placebo group, 18% in the OCA 10 mg group (p=0·045), and 23% in the OCA 25 mg group (p=0·0002)
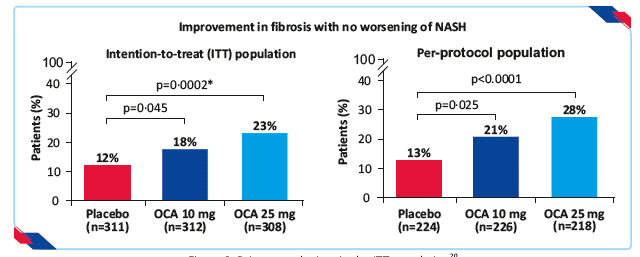
Figure 9: Primary endpoints in the ITT population20
- Dose-dependent response was observed in the ITT population, with more patients in the OCA 25 mg group showing at least a 1-point improvement in scores in key histological features of NASH compared with the placebo group (44% patients’ vs 36% for lobular inflammation [p=0·032] and 35% vs 23% for hepatocellular ballooning [p=0·0011] respectively)
- Dose-dependent response was observed in the ITT population, with more patients in the OCA 25 mg group showing at least a 1-point improvement in scores in key histological features of NASH compared with the placebo group (44% patients’ vs 36% for lobular inflammation [p=0·032] and 35% vs 23% for hepatocellular ballooning [p=0·0011] respectively)
- Greater number of patients on OCA 10 mg (21%) and OCA 25 mg (27%) exhibited improvement of fibrosis of at least one stage or resolution of NASH, without worsening of either compared to (16%) patients in the placebo group
- Significantly higher proportion of patients showed improvement in NAS by at least two points with no worsening of fibrosis, had no disease progression as assessed by no worsening of fibrosis and no worsening of NASH, and had improvement in fibrosis of at least two stages, in OCA 25 mg group as compared with placebo group
OCA 25 mg significantly improved fibrosis and key components of NASH disease activity among patients with NASH.20
OCA in Patients with Inadequate Response to UDCA: POISE Trial
Nevens et al., conducted a 12-month, double-blind, placebo-controlled, phase 3 trial to assess the longer-term efficacy, safety, and adverse-event profile of OCA in patients with PBC who were receiving daily doses of 5 mg or 10 mg. 217 patients who had an inadequate response to ursodiol or who found the side effects of ursodiol unacceptable were randomly assigned to receive OCA at a dose of 10 mg (the 10-mg group), OCA at a dose of 5 mg with adjustment to 10 mg if applicable (the 5–10-mg group), or placebo. The primary end point was an alkaline phosphatase level of less than 1.67 times the upper limit of the normal range, with a reduction of at least 15% from baseline, and a normal total bilirubin level.21
Results observed were:21
- The 5–10-mg group (46%) and 10-mg group (47%) showed higher rate of primary end point than the placebo group (10%) at month 12 (p<0.001 for both comparisons)
- Response to OCA was rapid, with a significant difference observed between each OCA group and the placebo group by week 2 and at each time point (p<0.001 for all comparisons)
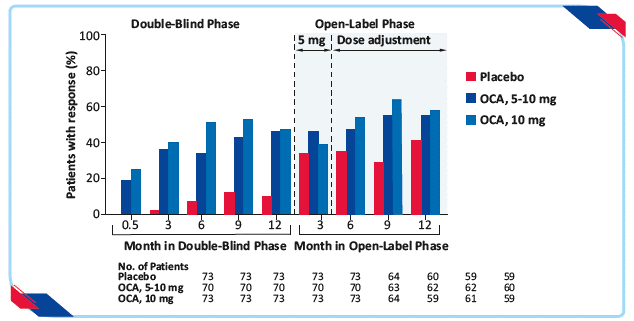
Figure 10: Primary composite end point in the double-blind and open-label extension phases, according to trial group21
- 5–10-mg OCA group and 10-mg OCA group showed significant reduction in the alkaline phosphatase level (least-squares mean, −113 and −130 U/L, resp, vs. −14 U/L; p<0.001 for both comparisons) as compared to the placebo group
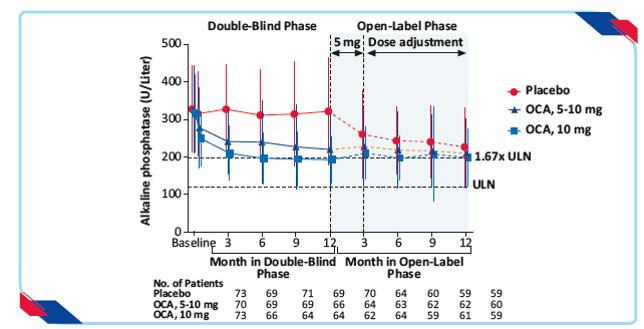
Figure 11: Alkaline phosphatase in the double blind and open-label extension phases, according to trial group21
- % of patients who had a reduction of at least 15% from baseline in the alkaline phosphatase level was higher in the 5–10-mg group (77%) and in the 10-mg group (77%) than in the placebo group (29%) (p<0.001 for both comparisons)
- OCA group showed significant reduction in the total bilirubin level as compared with a progressive increase in the placebo group (least-squares mean, −0.02±0.04 mg/dL [−0.3±0.7 μmol/L] in the 5–10-mg group and −0.05±0.04 mg/dL [−0.9±0.7 μmol/L] in the 10-mg group vs. 0.12±0.04 mg/dL [2.0±0.7 μmol/L] in the placebo group; p<0.001 for both comparisons)
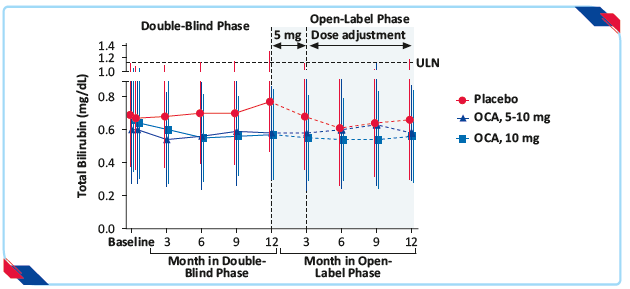
Figure 12: Total bilirubin levels in the double blind and open-label extension phases, according to trial group21
OCA administered with ursodiol or as monotherapy for 12 months in patients with primary biliary cholangitis resulted in decreases from baseline in alkaline phosphatase and total bilirubin levels that differed significantly from the changes observed with placebo.21
OBIHOLIC: Safety profile
The most common adverse reactions associated with OCA (≥5%) are: pruritus, fatigue, abdominal pain and discomfort, rash, oropharyngeal pain, dizziness, constipation, arthralgia, thyroid function abnormality, and eczema. The most common adverse reactions in Trial 1 occurring in at least 5% of patients in either OCA treatment arm and at an incidence at least 1% higher than the placebo treatment arm are shown in
table 1.16
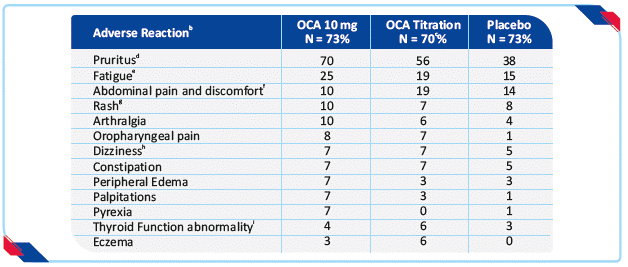
In the trial there were 16 patients (7%) who were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCA 10 mg arm, 5 patients (7%) in the OCA titration arm, and 5 patients (7%) in the placebo arm. b Occurring in greater than or equal to 5% of patients in either OCA treatment arm and at an incidence greater than or equal to1% higher than in the placebo treatment arm. Page 10 c Patients randomized to OCA titration received OCA 5 mg once daily for the initial 6 month period. At Month 6, patients who were tolerating OCA, but had an ALP 1.67-times ULN or greater, and/or total bilirubin greater than ULN, or less than 15% ALP reduction were eligible for titration from 5 mg once daily to 10 mg once daily for the final 6 months of the trial. d Includes skin eruptions, prurigo, pruritus, pruritus generalized, eye pruritus, ear pruritus, anal pruritus, vulvovaginal pruritus, and rash pruritic. e Includes fatigue, tiredness and asthenia. f Includes abdominal pain upper, abdominal pain, abdominal discomfort, abdominal pain lower, abdominal tenderness, and gastrointestinal pain. g Includes urticaria, rash, rash macular, rash papular, rash maculo-papular, heat rash, urticaria cholinergic. h Includes dizziness, syncope, presyncope. i Includes thyroxine free decreased, blood thyroid stimulating hormone increased, hypothyroidism.
Table 1: Most common adverse reactions in adult patients with PBC in trial 1 by treatment arm with or without UDCAa16
Full Prescribing Information
Therapeutic Indication16
OCA is indicated for the treatment of PBC in combination with UDCA in adults with an inadequate response to UDCA, or as monotherapy in adults unable to tolerate UDCA.
Dosage and Administration16
Prior to the initiation of OCA in patients with suspected cirrhosis, use the nomogram to calculate the patient’s score to determine their Child-Pugh classification (A, B, or C) and determine the appropriate starting dosage.16 Routinely monitor patients during OCA treatment for biochemical response, tolerability, progression of PBC disease, and re-evaluate Child-Pugh classification to determine if dosage adjustment is needed.16 Reduce the dosing frequency from once daily to once weekly as appropriate for patients who progress to advanced disease (i.e., from Child-Pugh Class A to Child-Pugh Class B or C).16The recommended starting dose and titration dosage regimen of OCA for patients who have not achieved an adequate biochemical response to an appropriate dosage of UDCA for at least 1 year or are intolerant to UDCA is dependent upon disease stage as shown in table below.
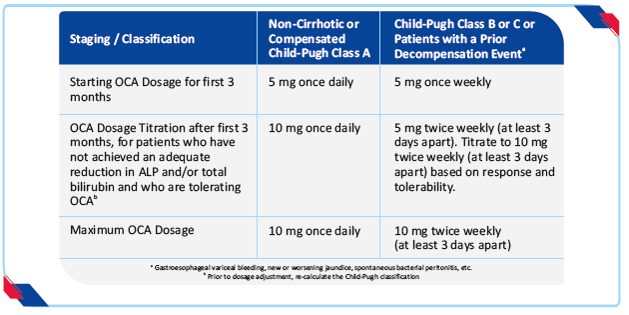
Table 2: Dosage regimen by disease stage16
- Non-cirrhotic patients or compensated cirrhotic patients with no or mild hepatic impairment (Child-Pugh Class A) are dosed once daily
- Cirrhotic patients with moderate or severe hepatic impairment (Child-Pugh Class B or C) or patients who have previously experienced a decompensation event are dosed initially once weekly and not more than twice weekly
Contra-indications16
OCA is contraindicated in patients with complete biliary obstruction.
Warnings and Precautions16
Hepatic decompensation and failure in incorrectly dosed PBC patients with Child-Pugh Class b or c or decompensated cirrhosis
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in PBC patients with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCA was dosed more frequently than the recommended starting dosage of 5 mg once weekly. Reported cases typically occurred within 2 to 5 weeks after starting OCA and were characterized by an acute increase in total bilirubin and/or ALP concentrations in association with clinical signs and symptoms of hepatic decompensation (e.g., ascites, jaundice, gastrointestinal bleeding, worsening of hepatic encephalopathy). A few cases reported improvement after OCA discontinuation; however, some cases reported ongoing symptoms. Because postmarketing cases often contain limited clinical information, there was insufficient information to rule out confounding factors (e.g., concomitant medications) or the role of the patient’s underlying advanced disease in the events. Patients who died due to liver-related complications generally had decompensated cirrhosis prior to treatment and were started on OCA 5 mg once daily, which is 7-fold greater than the once weekly starting regimen in this population
Liver-related adverse reactions16
In two 3-month, placebo-controlled clinical trials in patients with primarily early stage PBC disease, a dose-response relationship was observed for the occurrence of liver-related adverse reactions including jaundice, worsening ascites and primary biliary cholangitis flare with dosages of OCA of 10 mg once daily to 50 mg once daily (up to 5-times the highest recommended dosage), as early as one month after starting treatment with OCA. In a pooled analysis of three placebo-controlled trials in patients with primarily early stage PBC disease, the exposure-adjusted incidence rates for all serious and otherwise clinically significant liver-related adverse reactions, and isolated elevations in liver biochemical tests, per 100 patient exposure years (PEY) were: 5.2 in the OCA 10 mg group (highest recommended dosage), 19.8 in the OCA 25 mg group (2.5 times the highest recommended dosage) and 54.5 in the OCA 50 mg group (5 times the highest recommended dosage) compared to 2.4 in the placebo group. Monitor patients during treatment with OCA for elevations in liver biochemical tests and for the development of liver-related adverse reactions. Close monitoring is recommended for patients at an increased risk of hepatic decompensation. Interrupt treatment with OCA in patients with laboratory or clinical evidence of worsening liver function indicating risk of decompensation, and monitor the patient’s liver function.
Severe Pruritus16
Severe pruritus was reported in 23% of patients in the OCA 10 mg arm, 19% of patients in the OCA titration arm, and 7% of patients in the placebo arm in Trial 1, a 12-month double-blind randomized controlled trial of 216 patients. Severe pruritus was defined as intense or widespread itching, interfering with activities of daily living, or causing severe sleep disturbance, or intolerable discomfort, and typically requiring medical interventions. In the subgroup of patients in the OCA titration arm who increased their dosage from 5 mg once daily to 10 mg once daily after 6 months of treatment (n=33), the incidence of severe pruritus was 0% from Months 0 to 6 Page 8 and 15% from Months 6 to 12. The median time to onset of severe pruritus was 11, 158, and 75 days for patients in the OCA 10 mg, OCA titration, and placebo arms, respectively. Consider clinical evaluation of patients with new onset or worsening severe pruritus. Management strategies include the addition of bile acid resins or antihistamines, OCA dosage reduction, and/or temporary interruption of OCA dosing.
Reduction in HDL-C16
Patients with PBC generally exhibit hyperlipidemia characterized by a significant elevation in total cholesterol primarily due to increased levels of highdensity lipoprotein-cholesterol (HDL-C). In Trial 1, dose-dependent reductions from baseline in mean HDL-C levels were observed at 2 weeks in OCA-treated patients, 20% and 9% in the 10 mg and titration arms, respectively, compared to 2% in the placebo arm. At month 12, the reduction from baseline in mean HDL-C level was 19% in the OCA 10 mg arm, 12% in the OCA titration arm, and 2% in the placebo arm. Nine patients in the OCA 10 mg arm, 6 patients in the OCA titration arm, versus 3 patients in the placebo arm had reductions in HDL-C to less than 40 mg/dL. Monitor patients for changes in serum lipid levels during treatment. For patients who do not respond to OCA after 1 year at the highest recommended dosage that can be tolerated (maximum of 10 mg once daily), and who experience a reduction in HDL-C, weigh the potential risks against the benefits of continuing treatment.
Drug Interactions16
Bile Acid Binding Resins
- Bile acid binding resins such as cholestyramine, colestipol, or colesevelam adsorb and reduce bile acid absorption and may reduce the absorption, systemic exposure, and efficacy of OCA.
- If taking a bile acid binding resin, take OCA at least 4 hours before or 4 hours after taking the bile acid binding resin, or at as great an interval as possible.
Warfarin
- The International Normalized Ratio (INR) decreased following coadministration of warfarin and OCA.
- Monitor INR and adjust the dosage of warfarin, as needed, to maintain the target INR range when co-administering OCA and warfarin.
CYP1A2 Substrates with Narrow Therapeutic Index
- OCA may increase the exposure to concomitant drugs that are CYP1A2 substrates.
- Therapeutic monitoring of CYP1A2 substrates with a narrow therapeutic index (e.g. theophylline and tizanidine) is recommended when co-administered with OCA.
Inhibitors of Bile Salt Efflux Pump
- Avoid concomitant use of inhibitors of the bile salt efflux pump (BSEP) such as cyclosporine.
- Concomitant medications that inhibit canalicular membrane bile acid transporters such as the BSEP may exacerbate accumulation of conjugated bile salts including taurine conjugate of OCA in the liver and result in clinical symptoms. If concomitant use is deemed necessary, monitor serum transaminases and bilirubin.
Use in Specific Population16
Pregnancy Risk
The limited available human data on the use of OCA during pregnancy are not sufficient to inform a drug-associated risk. In animal reproduction studies, no developmental abnormalities or fetal harm was observed when pregnant rats or rabbits were administered OCA during the period of organogenesis at exposures approximately 13 times and 6 times human exposures, respectively, at the maximum recommended human dose (MRHD) of 10 mg. The estimated background risks of major birth defects and miscarriage for the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Lactation
There is no information on the presence of OCA in human milk, the effects on the breast-fed infant or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for OCA and any potential adverse effects on the breastfed infant from OCA or from the underlying maternal condition.
Pediatric Use
The safety and effectiveness of OCA in pediatric patients have not been established.
Geriatric Use
Of the 201 patients in clinical trials of OCA who received the recommended dosage (5 mg or 10 mg once daily), 41 (20%) were 65 years of age and older, while 9 (4%) were 75 years of age and older. No overall differences in safety or effectiveness were observed between these subjects and subjects less than 65 years of age, but greater sensitivity of some older individuals cannot be ruled out.
Hepatic Impairment
Hepatic decompensation and failure, in some cases fatal, have been reported postmarketing in PBC patients with decompensated cirrhosis or Child-Pugh B or C hepatic impairment when OCA was dosed more frequently than recommended. In PBC clinical trials, a dose-response relationship was observed for the occurrence of liver-related adverse reactions with OCA. Plasma exposure to OCA and its active conjugates, increases significantly in patients with moderate to severe hepatic impairment (Child-Pugh Classes B and C). Prior to the initiation of OCA determine the patient’s Child-Pugh classification to determine the starting dosage. Re-evaluate the dosing regimen periodically during treatment. Dosage adjustment is required in patients with Child-Pugh Class B and C. Routinely monitor patients for progression of PBC disease with laboratory and clinical assessments. Dosage adjustment, interruption or discontinuation may be required.
Key Highlights
- The proven first-line treatment and the current standard of care for PBC is Ursodeoxycholic acid
- However, up to 40 % of patients have an incomplete biochemical response to UDCA, and the risk of progression to end-stage liver disease and worse transplant-free survival rates have been shown to be correlated with this than UDCA-responsive patients
- Additional treatment options are also required for this category of patients to enhance clinical outcomes
- OCA is the first FXR agonist approved by the FDA that reduces bile acid synthesis and PBC-induced hepatic damage
- This new mechanism of action offers an alternative treatment for patients who are unable to tolerate UDCA or can be used to suppress ALP and other biochemical markers as an additive therapy
References
- Dharmalingam M, Yamas PG. Nonalcoholic fatty liver disease and Type 2 diabetes mellitus. Indian J Endocr Metab 2018;22:421-8.
- Tomah S, Alkhouri N, Hamdy O. Nonalcoholic fatty liver disease and type 2 diabetes: where do Diabetologists stand? Clin Diabetes Endocrinol. 2020;6:9.
- Godoy-Matos AF, Silva Júnior WS, Valerio CM. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol Metab Syndr. 2020 Jul 14;12:60.
- Gastaldelli A, Cusi K. From NASH to diabetes and from diabetes to NASH: Mechanisms and treatment options. JHEP Rep. 2019 Jul 19;1(4):312-328.
- Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018 Jan;67(1):328-357.
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016 Jun;64(6):1388-402.
- American Diabetes Association. 4. Comprehensive Medical Evaluation and Assessment of Comorbidities: Standards of Medical Care in Diabetes-2020. Diabetes Care. 2020 Jan;43(Suppl 1):S37-S47.
- Navarro R. Challenges and opportunities in the management of advanced fibrosis due to nonalcoholic steatohepatitis (NASH). https://cdn.sanity.io/files/0vv8moc6/ajmc/cebfd6c1801aacc915ae1dbf2d3af8ea0933c38f.pdf
- Corpechot C, Poupon R, Chazouillères O. New treatments/targets for primary biliary cholangitis. JHEP Rep. 2019;1(3):203-213.
- Zimmer V, Lammert F. Looking Into the Crystal Ball: Predicting Non-response to Ursodeoxycholic Acid in Primary Biliary Cholangitis. EBioMedicine. 2017;15:10-11.
- Han CY. Update on FXR Biology: Promising Therapeutic Target?. Int J Mol Sci. 2018;19(7):2069.
- Ratziu V, Sanyal AJ, Loomba R, Rinella M, Harrison S, Anstee QM, et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp Clin Trials. 2019 Sep;84:105803.
- Smith SM, Pegram AH. Obeticholic Acid A Farnesoid X Receptor Agonist for Primary Biliary Cholangitis. J Pharm Technol. 2017 Apr; 33(2): 66–71.
- Chapman RW, Lynch KD. Obeticholic acid-a new therapy in PBC and NASH. Br Med Bull. 2020 May 15;133(1):95-104.
- Markham A, Keam SJ. Obeticholic Acid: First Global Approval. Drugs. 2016 Aug;76(12):1221-6.
- OCA Prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207999s003lbl.pdf (Last accessed November 2020).
- Khanna A, Jones DE. Novel strategies and therapeutic options for the management of primary biliary cholangitis. Therap Adv Gastroenterol. 2017;10(10):791‐
- Assessment report. OCALIVA. Available at: https://www.ema.europa.eu/en/documents/assessment-report/ocaliva-epar-public-assessment-report_en.pdf (Last accessed November 2020).
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Natta MLV, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015 Mar 14;385(9972):956-65.
- Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al; REGENERATE Study Investigators. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019 Dec 14;394(10215):2184-2196.
- Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N Engl J Med. 2016 Aug 18;375(7):631-43.
