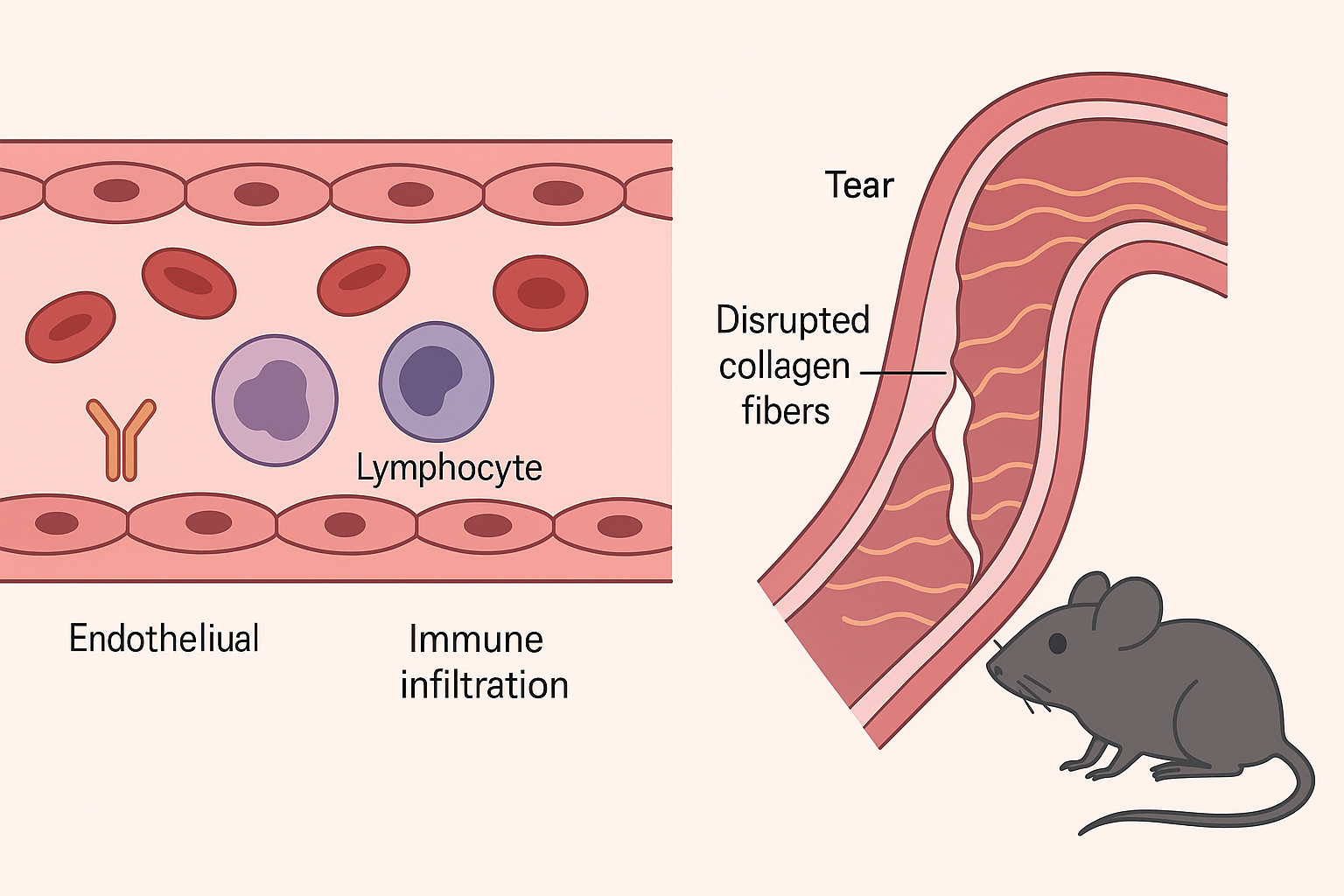
The study presents a novel mouse model for spontaneous aortic dissection (AD), developed by introducing a patient-derived FBN1 gene variant (Gly234Asp) using CRISPR/Cas9. This mutation, identified in a family with nonsyndromic AD, leads to early-onset, rapidly progressing aortic dissection in homozygous mutant mice, with a 50% mortality rate by five weeks of age. The mutant mice exhibit multiple intimomedial tears in the ascending aorta, mimicking human pathology. The research identifies early abnormalities in endothelial cells (ECs), such as misalignment with blood flow and upregulation of adhesion molecules (VCAM-1 and ICAM-1), contributing to immune cell recruitment.
Histological, imaging, and single-cell RNA sequencing analyses reveal the accumulation of monocytes and macrophages in the intima before the onset of dissection. These immune cells display both M1- and M2-like characteristics and are associated with increased expression of inflammatory markers and matrix metalloproteinases (MMP2/9), indicating active tissue remodeling and inflammation. Additionally, the mutant FBN1 protein loses its binding ability to latent transforming growth factor-beta binding proteins (LTBP-1, -2, and -4), leading to suppressed TGFβ signaling—a notable departure from other Marfan syndrome models where TGFβ signaling is typically upregulated.
The study demonstrates that the loss of FBN1 function impairs extracellular matrix integrity and elastogenesis in smooth muscle cells, fostering a permissive environment for AD development. Endothelial dysfunction emerges as an early trigger, with inflammatory cell infiltration and compromised vascular wall structure driving disease progression. Attempts to deplete macrophages using clodronate liposomes reduced immune cell presence but did not prevent dissection or restore tissue integrity.
This model provides crucial insights into the interplay between structural matrix defects, immune responses, and endothelial signaling in AD. It underscores the importance of TGFβ signaling regulation by FBN1 and offers a valuable platform for exploring targeted therapies against inflammation-driven aortic pathology.
Source: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.125.326230
